发布时间:2025-05-16 人气:1334 作者:天之恒
美国食品药品监督管理局(FDA)对医疗器械的监管体系以科学严谨著称,其注册认证流程的周期与费用因产品风险等级、技术复杂程度及申请路径选择而呈现显著
差异。本文将从流程周期、费用构成及影响因素三个维度展开分析,为企业提供精准的决策参考。
一、注册认证周期:风险分级与路径选择决定时效
FDA将医疗器械分为Ⅰ、Ⅱ、Ⅲ类,风险等级依次递增,注册周期随之延长。
Ⅰ类医疗器械(如手术刀、检查手套):通常采用豁免510(k)的路径,仅需完成企业注册和产品列名。若文件完整,最快可在1周内完成电子注册,部分简单产品甚至可
实现5-7个工作日的快速审批。
Ⅱ类医疗器械(如X光机、血糖仪):需提交510(k)预市通知,证明与已上市产品实质等同。该流程平均耗时6个月,但若涉及新型技术或补充材料,周期可能延长至9-12个月。
Ⅲ类医疗器械(如人工心脏、植入式除颤器):必须通过PMA(上市前批准)申请,需提交完整的临床试验数据。其审批周期长达10-12个月,复杂案例甚至超过18个月。
周期波动还受FDA审查负荷、政策调整及企业响应效率影响。例如,2025年FDA优化了电子提交系统,使部分Ⅱ类产品的技术评估阶段缩短约20%。
二、费用构成:官费、代理费与隐性成本并存
注册费用由三部分组成,且随财政年度动态调整。
FDA官费:
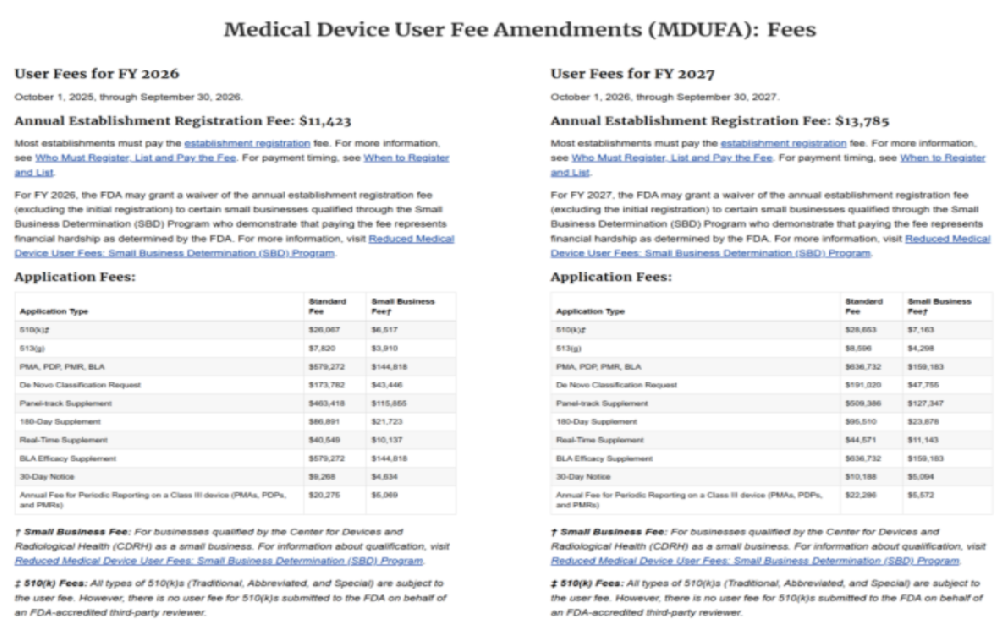
企业注册费:2025财年为9280美元,需每年缴纳以维持注册状态。
申请审核费:
510(k)申请:标准费用24,335美元,小企业(年收入≤1亿美元)可享优惠至6,084美元。
PMA申请:费用高达310,764美元,无小企业优惠。
代理服务费:
国内专业咨询机构收费因服务深度而异:
Ⅰ类:4,500-10,000元人民币(仅注册代理)。
Ⅱ类:7-12万元人民币(含510(k)文件编制)。
Ⅲ类:30-100万元人民币(含PMA全流程支持)。
隐性成本:
临床试验:Ⅲ类设备平均成本超500万美元,涉及受试者招募、数据管理等。
技术整改:若FDA要求补充测试或文件修订,可能产生额外费用。
三、关键影响因素:企业需前置规划
产品分类准确性:误判风险等级可能导致申请路径错误,如将Ⅱ类按Ⅰ类申报,将直接遭FDA拒收。
文件完整性:缺失临床前研究数据或生物相容性报告,可能触发30-90天的补充材料周期。
沟通效率:积极响应FDA提问可缩短审查时间。数据显示,Ⅱ类申请中,首次回复在15个工作日内的企业,平均获批周期缩短25%。
政策变动:如2025年FDA对人工智能医疗器械启动试点计划,相关产品可申请加速审查,周期压缩至4-6个月。
结语:科学规划与专业支持是关键
美国医疗器械注册认证是一项系统性工程,企业需结合产品特性、预算及上市计划,选择最优路径。建议通过以下步骤优化流程:
提前分类:使用FDA产品分类数据库(Product Classification Database)精准定位风险等级。
模拟预审:借助第三方机构进行文件模拟评审,降低补正风险。
动态跟踪:关注FDA官网(fda.gov)的财政年度费用公告及政策更新。
在全球化竞争中,合规效率直接决定市场先机。通过科学规划与专业协作,企业可有效平衡注册成本与时间成本,加速产品进入美国市场的进程。
















 客服1
客服1